AWK is awesome
AWK is awesome because it is the most rewarding programing language you can learn for bioinformatics and Data Science. There is little to learn and it is useful every day.
AWK has been the most beneficial programming language I have ever learned. It took me only a day to learn most of it and it saved me several weeks if not months already. I use AWK almost every day.
It is better to see AWK in action once than to hear about it a thousand times. So, let’s start with the examples.
Table summary
I usually use AWK to calculate some simple summary statistics for a table. For example, let’s assume you have a file table.txt with some numeric values:
CHR STAR END LOD SCORE
chr1 211829 211850 lod=31 333
chr1 211867 211871 lod=13 247
chr1 211877 211903 lod=66 408
chr1 211913 211927 lod=61 400
chr1 211971 211994 lod=60 399
chr1 211996 212024 lod=72 417
chr6 310311 310324 lod=16 268
chr6 312061 312066 lod=13 247
chr6 312100 312206 lod=376 580
chr6 312653 312728 lod=19 285
chr6 312908 313028 lod=348 573
chr6 313549 313788 lod=900 667
chr6 313589 313784 lod=747 648
Mean
You can quickly get the mean SCORE value:
awk '{s+=$5} END {print s/(NR-1)}' table.txt

where s+=$5 sums up all values of the 5th column;
NR is a built-in variable that equals to the row number. I use NR-1 because I skip the header.
The command after the END is executed when the end of the file is reached.
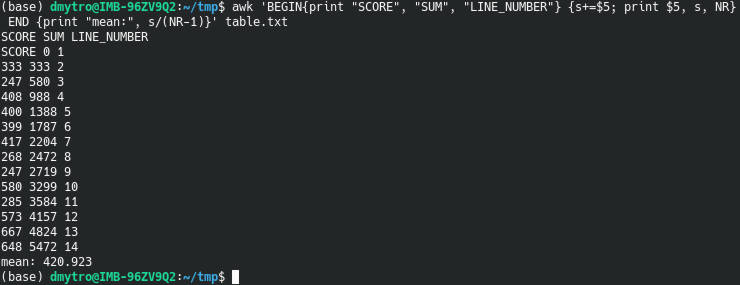
To see what AWK does line by line, run this command:
awk 'BEGIN{print "SCORE", "SUM", "LINE_NUMBER"} {s+=$5; print $5, s, NR} END {print "mean:", s/(NR-1)}' table.txt
But how to calculate the mean of the LOD columns that has lod= in front of each number?
You can use AWK to clean tha data and do the calculation:
awk 'gsub( "lod=", "" , $4){s+=$4}END{print s/(NR-1)}' table.txt

gsub( "lod=", "" , $4) replaces lod= with an empty string before any calculation is done.
You can also limit the calculation to one chromosome:
awk '$1=="chr1" {n++; s+=$5} END {print s/n}' table.txt

We use the condition if ($1=="chr1") do ({n++; s+=$5}). Also, NR is replaced with n++ to count only the lines that meet the condition $1=="chr1"
Min and max
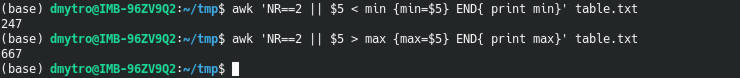
Using the same principles, you can get the minimum and maximum values of the SCORE column:
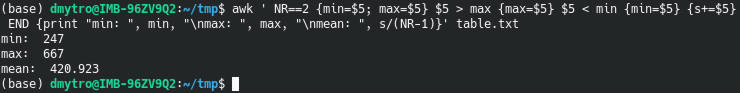
awk 'NR==2 || $5 < min {min=$5} END{ print min}' table.txt
awk 'NR==2 || $5 > max {max=$5} END{ print max}' table.txt
|| means OR statement in AWK.
You can combine these two commands in one:
awk 'NR==2 {min=$5; max=$5} $5 > max {max=$5} $5 < min {min=$5} END {print "min: ", min, "\nmax: ", max}' table.txt
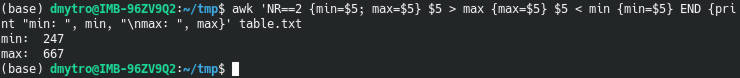
NR==2 {min=$5; max=$5} assigns the initial values of min and max using the second row.
$5 > max {max=$5} and $5 < min {min=$5} are conditional statements that are checked one after another.
Mean, max, and min in one line
You can also combine all three calculations in one line and get all statistics in one run:
awk ' NR==2 {min=$5; max=$5} $5 > max {max=$5} $5 < min {min=$5} {s+=$5} END {print "min: ", min, "\nmax: ", max, "\nmean: ", s/(NR-1)}' table.txt
Genotypes summary
There are more complicated cases where you can use AWK.
You may want to do some calculations of the genotype table generated by VariantsToTable from the GATK:
#CHROM POS REF 12.4.GT 13.16.GT 16.9.GT
scaffold_1 191 A ./. ./. A/A
scaffold_1 563 T T/T ./. T/A
scaffold_1 647 A C/C C/C A/C
scaffold_1 669 T T/T T/T T/T
scaffold_1 679 C C/A C/A C/A
scaffold_1 704 T T/C T/C T/C
scaffold_1 721 T C/C C/C C/C
scaffold_1 722 C C/T C/T C/T
scaffold_1 733 G G/T G/T G/*
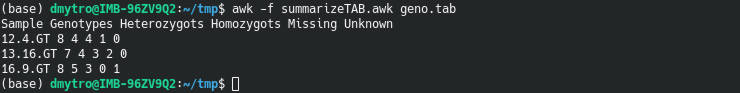
For example, I often calculate the number heterozygous, homozygous sites and missing genotypes. To that end, I use this AWK script written in the summarizeTAB.awk file:
{if (NF > maxNF ) {
for (i = 4; i <= NF; i++)
countN[i] = 0; countHomo[i] = 0; countHetero[i] = 0; countNA[i] = 0; maxNF = NF;
}
if (NR == 1 ) { for (i = 4; i <= NF; i++) samples[i] = $i;}
else {
for (i = 3; i <= NF; i++)
{if ($i == "N" || $i == "./.") countN[i]++;
else if ($i == "A/A" || $i == "T/T" || $i == "G/G" || $i == "C/C") countHomo[i]++;
else if ($i == "G/A" || $i == "T/C" || $i == "A/C" || $i == "G/T" || $i == "C/G" || $i == "A/T" || \
$i == "A/G" || $i == "C/T" || $i == "C/A" || $i == "T/G" || $i == "G/C" || $i == "T/A") \
countHetero[i]++;
else countNA[i]++;
}
}
}
END {
print "Sample", "Genotypes", "Heterozygots", "Homozygots", "Missing", "Unknown";
for (i = 4; i <= maxNF; i++)
print samples[i], countHomo[i]+countHetero[i]+0, countHetero[i]+0, countHomo[i]+0, countN[i]+0, countNA[i]+0;
}
It loops through the columns starting from the 4th one and calculates the number of the number heterozygous, homozygous, missing, and unknown genotypes. These number are stored in corresponding variables.
When the script is too long to fit it in one line as in this case, you can write it into a file and tell AWK to execute it:
awk -f summarizeTAB.awk geno.tab
AWK vs Python
The AWK code is usually shorter and works faster than Python. I do not have a dramatic example when my AWK code is substantially shorter than the equivalent Python code. But there are great examples from other AWK users.
Be careful with AWK
There is one key point you need to keep in mind when you work with AWK. It doesn’t throw an error when it encounters something unusual. Instead, AWK tried to guess how to handle it and proceeds silently. This can put you in danger.
Using the mean SCORE column example from above, you can see that AWK treated the characters in the header as 0.
This would throw you an error in Python. A character string and numeric value cannot be summed. But AWK doesn’t give such an error.
You would have done a mistake in the mean calculation if you calculated the line numbers as n++. It would have counted the header too. That’s why I also deduced 1 from the number of rows specified with NR.
Similarly, if you have missing data points in a form of NA, you need to tell AWK to skip them:
awk 'NR>1 && $5!="NA" {s+=$5; n++; print $5, s, n} END {print "mean:", s/n}' table.txt
I also used NR>1 to skip the header.
So, you need to be aware of this behavior of AWK when you have a mixture of data types.
Where to learn AWK
If you want to learn AWK, I recommend the course “To awk or not to…”. It was fantastic when I took it in 2017, and it has improved since then.
I also often visit this AWK page, for quick reference on the functions.
If you have never used AWK, give it a try. It may change your life forever.