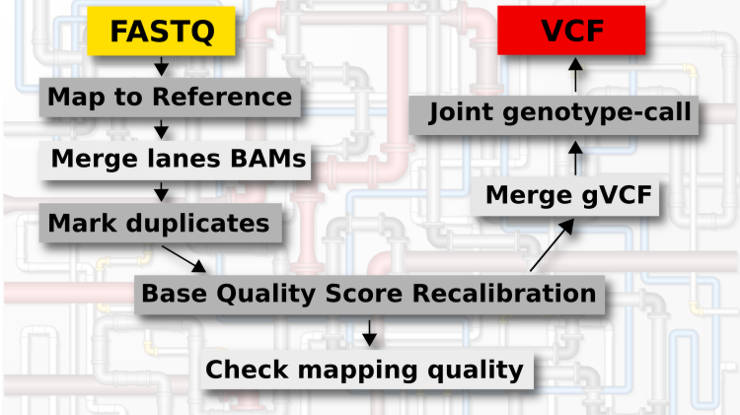
Genomic variant calling pipeline
An automated genomic variant calling pipeline becomes essential when a project scales to hundreds of genomes. Here is my genomic variant calling pipeline.
As probably any beginner, I used to process my genomic data with manual interference at every step. So, I would submit mapping jobs for all samples on a computing cluster, when they all done I would submit mark duplicates jobs, etc. Moreover, I would also manually write sbatch scripts (my cluster UPPMAX uses the Slurm Workload Manager). It was not efficient.
Well, I used replacements (with sed) and loops (with for i in x; do ...) to reduce the amount of work, but there were many manual steps. I managed to process 24-31 small Capsella genomes (~200Mb) this way during my PhD projects. Now, I work with the dog genome which is much bigger (~2.5Gb) and I also need to analyze many more samples (82 genomes at the moment). So, I had to write this genomic variant calling pipeline to make my workflow as automatic as possible.
This genomic variant calling pipeline was written for the dog data, but you can modify it for any other organism with a reference genome (see below). If you work with non-model organisms, I recommend you also check my GATK tutorial for non-model organisms.
Requirements
For this genomic variant calling pipeline you would need the following software installed:
- BWA or Stampy - mapping.
- Samtools - SAM/BAM files manipulation.
- Picard - mark duplicates.
- QualiMap (optional) - check the mapping quality.
- GATK - genotype calling.
Genomic variant calling pipeline
Main steps
This genomic variant calling pipeline includes the following steps:
- Mapping to the reference.
- Merging BAM files of different lanes.
- Mark duplicates.
- Base Quality Score Recalibration (BQSR).
- Check mapping quality (optional).
- Genotype each sample in the GVCF mode.
- Merge gVCF files from different samples.
- Joint genotyping of all samples.
How to use
This genomic variant calling pipeline consists of two files reads-to-VCF_dog.py (main script) and sbatch.py (all the functions) that can be downloaded from my Github repository.
To run it, you need a list of FASTQ files with R1 only names. You can obtain such a list by listing the content of the folder with your FASTQ files:
ls -l fastq/*.fastq.gz | grep R1 | sed 's/ \+/ /g' | cut -d " " -f 9 > R1.txt
Next, you run the reads-to-VCF.py script using R1.txt as an input.
python reads-to-VCF.py -i R1.txt -r canFam3.fa -p snic2017 -n 20 -t 5-00:00:00 -l \$SNIC_TMP -a BWA -m 4 -k "dogs.557.publicSamples.ann.chrAll.PASS.vcf.gz,00-All_chrAll.vcf.gz"
You can see the description of each option by running:
python reads-to-VCF.py -h
I only would like to mention that you can use either BWA or stampy aligners for the mapping step (option -a). In case of stampy, you can also specify the per base pair divergence from the reference (-d).
Among other options, snic2017 is the project name that is needed for the Slurm on Uppmax, I am not sure if you need it in other systems.
I also recommend providing the maximum available number of cores (-n) for parallel jobs, and maximum available RAM per core (-m).
I also request 5 days (-t) for each main step to run on a cluster.
For many intermediate steps, I make use of the temporary storage $SNIC_TMP (you need to add \ before the $ sign because it is a special character). This helps to keep my account disk use low. Moreover, it makes the pipeline more efficient because $SNIC_TMP is located on the same node where the job is running. You need to provide your temporary directory with the option -l.
Assuming the content of R1.txt is the following
WCRO84_S23_L005_R1_test.fastq.gz
WCRO84_S23_L006_R1_test.fastq.gz
WCRO86_S21_L005_R1_test.fastq.gz
WCRO86_S21_L007_R1_test.fastq.gz
The script reads-to-VCF.py will generate these files:
GVCF_all.sbatch
WCRO84_L005_map.sh
WCRO84_L006_map.sh
WCRO84_gVCF.sh
WCRO84_mergeMarkDuplBQSR.sh
WCRO84_qualimap.sh
WCRO86_L005_map.sh
WCRO86_L007_map.sh
WCRO86_gVCF.sh
WCRO86_mergeMarkDuplBQSR.sh
WCRO86_qualimap.sh
reads-to-VCF_wolf.sbatch
All *.sh files contain commands to execute jobs. The file is reads-to-VCF.sbatch contains the BASH script to submit these jobs to the cluster with the dependencies between jobs.
You place all these files in the same directory and submit all the jobs with:
sh reads-to-VCF.sbatch
If everything worked fine, you will see this output:
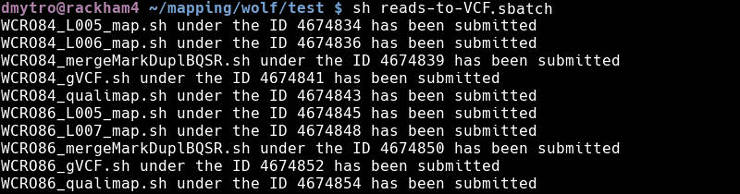
If you check the submitted jobs (with jobinfo), you will see the job dependencies (in red):
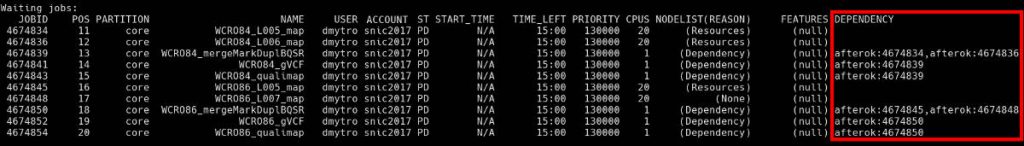
Following the job IDs, you can see that, for instance, the job WCRO84_mergeMarkDuplBQSR won't start until the two mapping jobs WCRO84_L005_map and WCRO84_L006_map are not finished successfully. When all these jobs finish, you will get a set of gVCF files, which you can merge and jointly genotype with GVCF_all.sbatch:
sbatch GVCF_all.sbatch
Thus, you run only 4 commands and you get from a set if FASTQ files to one VCF file.
I do not make the last command to execute automatically because it depends on all samples in this genomic variant calling pipeline. If your pipeline runs on many samples it is very likely that some samples will fail simply due to chance. For example, when I run this pipeline on 82 samples, 3 of them failed due to node failure.
If some of the jobs fail, the jobs dependent on it will be canceled.
To check if any job failed, run (assuming you use Slurm):
finishedjobinfo | grep FAILED
Then investigate why it failed. If it is not your mistake and it failed due to node failure, restart failed jobs by extracting and executing the code for the failed jobs from reads-to-VCF.sbatch.
I also suggest to list all the final BAM and gVCF files and check their size before executing GVCF_all.sbatch:
ls -l *.gz *.bam
If some samples have extremely large or extremely small files, check their *.fastq, *.sh *.err and *.out files to figure out what the problem is.
Also, you may want to modify GVCF_all.sbatch for your needs. For example, you may want to genotype mitochondrial and sex chromosomes with the control of ploidy (e.g. -ploidy 1 -L chrX -L chrM).
Generally, there should not be any problem and you will get your VCF file with as little effort as 4 commands.
Note, this genomic variant calling pipeline was designed and tested on UPPMAX with the Slurm Workload Manager, if you run it on another cluster, you may need to modify it. Below, I provide more details for each step and describe the hardcoded parts.
Description of each step
1. Mapping to the reference
The very first step of this genomic variant calling pipeline is to map all reads to the reference genome. Many of my samples were sequenced on different lanes. So, to control for this factor and to make the mapping process faster, I perform the mapping of each lane separately. An example command of the BWA mapping with subsequent sorting and converting to BAM by Samtools looks like this:
bwa mem -t 20 -M -R '@RG\tID:WCRO84_L005\tPL:illumina\tLB:WCRO84\tSM:WCRO84' canFam3.fa WCRO84_S23_L005_R1_test.fastq.gz WCRO84_S23_L005_R2_test.fastq.gz | samtools sort -O bam -@ 20 -m 4G > WCRO84_L005.bam
where WCRO84 is the sample name, and L005 is the lane number. These names are changed by the reads-to-VCF.py script. You can read about the options of BWA and Samtools in the documentation of these programs.
This step can be parallelized and I use 20 threads here.
If you choose to map with Stampy, the mapping command will look like this:
stampy.py -g canFam3 -h canFam3 --substitutionrate=0.002 -t 20 -o $SNIC_TMP/WCRO84_L005_stampy.bam --readgroup=ID:WCRO84_L005 -M WCRO84_S23_L005_R1_test.fastq.gz WCRO84_S23_L005_R2_test.fastq.gz
samtools sort -O bam -@ 20 -m 4G $SNIC_TMP/WCRO84_L005_stampy.bam > $SNIC_TMP/WCRO84_L005.bam
java -Xmx4G -jar picard.jar AddOrReplaceReadGroups \
I=$SNIC_TMP/WCRO84_S23_L005.bam \
O=WCRO84_S23_L005.bam \
RGID=WCRO84_S23_L005 \
RGLB=WCRO84 \
RGPL=illumina \
RGPU=WCRO84_S23_L005 \
RGSM=WCRO84
The last command adds read-group tags. I was not able to past these tags correctly with --read-group option in stampy. So, I added this extra step.
Mapping with Stampy is preferable if you map reads that are divergent from the reference. For example, I use Stampy to map wolf reads to the dog reference genome with the account for divergence from the reference (-d 0.002).
There are separate files generated for each mapping job. The example above would be named as WCRO84_L005_map.sh.
2. Merge lanes, mark duplicates, BQSR
All these three steps are performed as a sequence in one cluster job on a single core. So, they all are listed in one file, e.g. WCRO84_mergeMarkDuplBQSR.sh.
Merge lanes
The BAM files of different lanes are then merged for each samples:
ls WCRO84_L00?.bam | sed 's/ / /g;s/ /\n/g' > WCRO84_bam.list
samtools merge -b WCRO84_bam.list $SNIC_TMP/WCRO84_merged.bam
xargs -a WCRO84_bam.list rm
The first line creates a list of BAM files to merge. The second line performs merging. The last line removed the BAM files that have been merged to free some disk space. The file WCRO84_bam.list can be checked to make sure all the BAM files were included during the merging step.
Mark duplicates
This step identifies reads that are likely artificial duplicates originated during sequencing workflow. These reads are not removed but they will be ignored by default during the variant calling process.
java -Xmx4G -Djava.io.tmpdir=$SNIC_TMP -jar picard.jar MarkDuplicates \
VALIDATION_STRINGENCY=LENIENT \
METRICS_FILE=WCRO84_merged_markDupl_metrix.txt \
MAX_FILE_HANDLES_FOR_READ_ENDS_MAP=15000 \
INPUT=$SNIC_TMP/WCRO84_merged.bam \
OUTPUT=$SNIC_TMP/WCRO84_merged_markDupl.bam
rm $SNIC_TMP/WCRO84_merged.bam
The rm cleans unneeded files.
Base Quality Score Recalibration (BQSR)
This part is the slowest in the script. It applies some machine learning to detect and correct systematic errors in the base quality scores.
You need a good reference database of SNPs and indels for this step. I have used some of the files which I cannot share, but if you also run this genomic variant calling pipeline for dog data, you can download these two publicly available variant files:
wget ftp://ftp.ebi.ac.uk/pub/databases/eva/PRJEB24066/dogs.557.publicSamples.ann.vcf.gz*
wget ftp://ftp.ncbi.nih.gov/snp/organisms/archive/dog_9615/VCF/00-All.vcf.gz*
I also removed all filtered positions, because BQSR needs only the high confidence sites:
zcat 00-All.vcf | awk '{if (/^#/) {print $0} else {print "chr"$0}}' | bgzip > 00-All_chrAll.vcf
zcat dogs.557.publicSamples.ann.vcf.gz | awk '{if (/^#/) {print $0} else if ($7=="PASS") {print "chr"$0}}' | bgzip > dogs.557.publicSamples.ann.chrAll.PASS.vcf
tabix 00-All_chrAll.vcf & tabix dogs.557.publicSamples.ann.chrAll.PASS.vcf
To perform BQSR, you run these commands:
# Generate the first pass BQSR table file
gatk --java-options "-Xmx4G" BaseRecalibrator \
-R canFam3.fa \
-I $SNIC_TMP/WCRO84_merged_markDupl.bam \
--known-sites 00-All_chrAll.vcf.gz \
--known-sites dogs.557.publicSamples.ann.chrAll.PASS.vcf.gz \
-O WCRO84_merged_markDupl_BQSR.table
# Apply BQSR
gatk --java-options "-Xmx4G" ApplyBQSR \
-R canFam3.fa \
-I $SNIC_TMP/WCRO84_merged_markDupl.bam \
-bqsr WCRO84_merged_markDupl_BQSR.table \
-O WCRO84_merged_markDupl_BQSR.bam
rm $SNIC_TMP/WCRO84_merged_markDupl.bam
# Generate the second pass BQSR table file
gatk --java-options "-Xmx4G" BaseRecalibrator \
-R canFam3.fa \
-I WCRO84_merged_markDupl_BQSR.bam \
--known-sites 00-All_chrAll.vcf.gz \
--known-sites dogs.557.publicSamples.ann.chrAll.PASS.vcf.gz \
-O WCRO84_merged_markDupl_BQSR2.table
# Plot the recalibration results
gatk --java-options "-Xmx4G" AnalyzeCovariates \
-before WCRO84_merged_markDupl_BQSR.table \
-after WCRO84_merged_markDupl_BQSR2.table \
-plots WCRO84_merged_markDupl_BQSR.pdf
As I have mentioned it in my GATK: the best practice for non-model organisms, the BQSR can actually hurt if your variant reference database is not good enough. So, check the plots in WCRO84_merged_markDupl_BQSR.pdf to make sure your recalibration worked correctly. Mine worked fine and it looked like this:
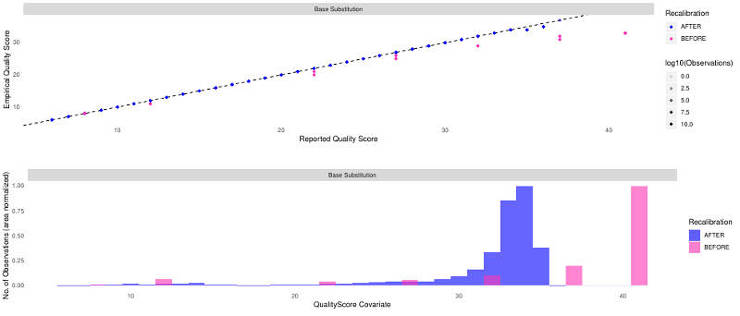
3. Check mapping quality (Optional)
This step is not essential for the genomic variant calling pipeline, but it useful to check the quality of your BAM files. With Qualimap you get a lot of useful information such as coverage, number of mapped/unmapped reads, mapping quality, etc. An example of the file name for this step is WCRO84_qualimap.sh.
The command is the following:
qualimap bamqc -nt 20 --java-mem-size=4G -bam WCRO84_merged_markDupl_BQSR.bam -outdir WCRO84_qualimap
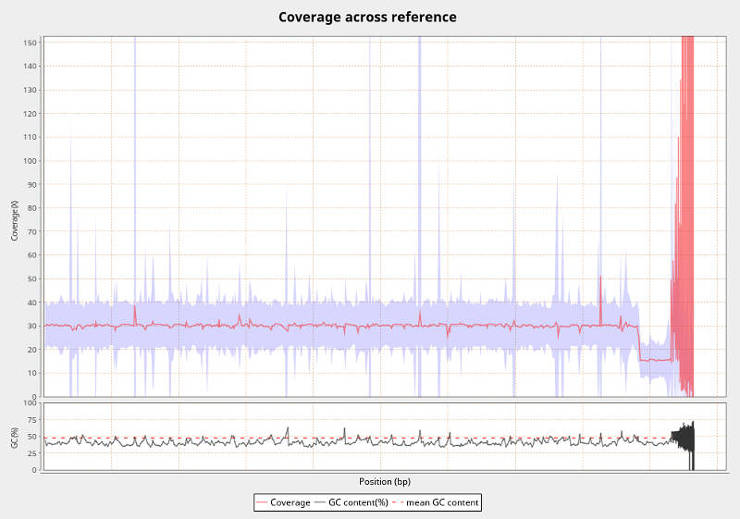
The report will be in an HTML file which you can view in your browser. For really many samples, you can also batch-process the TXT files with results. For example, to extract the mean coverage information, run
grep "mean coverageData" */genome_results.txt
At this stage, the work with BAM files is finished and the pipeline proceeds to the genotyping steps.
4. Call variants per-sample (gVCF)
This step runs the GATK HaplotypeCaller in GVCF mode:
gatk --java-options "-Xmx4G" HaplotypeCaller \
-R canFam3.fa \
-ERC GVCF \
-I WCRO84_merged_markDupl_BQSR.bam \
-O WCRO84_merged_markDupl_BQSR.g.vcf.gz
tabix WCRO84_merged_markDupl_BQSR.g.vcf.gz
mkdir WCRO84
mv WCRO84_* WCRO84
This enables to generate intermediate files, which are then used for joint genotyping in an efficient way. tabix is necessary to index the compressed file for the GATK compatibility in the next steps.
At this step, I also create sample specific directories and move all the files into the sample's own directory. This helps to keep the order when you work with many files.
An example of the file name for this step is WCRO84_gVCF.sh.
5. Joint genotyping of all samples.
This step merges all gVCF files from multiple samples:
gatk --java-options "-Xmx4g -DGATK_STACKTRACE_ON_USER_EXCEPTION=true" CombineGVCFs \
-R canFam3.fa \
-V WCRO84/WCRO84_merged_markDupl_BQSR.g.vcf.gz \
-V WCRO86/WCRO86_merged_markDupl_BQSR.g.vcf.gz \
-O GVCF_merged_markDupl_BQSR.g.vcf.gz
tabix GVCF_merged_markDupl_BQSR.g.vcf.gz
In the example command above, I provide only 2 samples to keep it simple. Obviously, there will be more samples in a real run.
The final command performs joint-call of SNPs and indels:
gatk --java-options "-Xmx4g -DGATK_STACKTRACE_ON_USER_EXCEPTION=true" GenotypeGVCFs \
-R reference/canFam3.fa \
-V GVCF_merged_markDupl_BQSR.g.vcf.gz \
-O GVCF_merged_markDupl_BQSR.vcf.gz
tabix GVCF_merged_markDupl_BQSR.vcf.gz
The file name of the script that includes both these commands is GVCF_all.sbatch.
Script to submit all these jobs
All the job described above are submitted by executing this script (2 samples example):
#!/bin/sh
WCRO84_L005=$(sbatch -A snic2017 -p core -n 20 -t 5-00:00:00 -J WCRO84_L005_map -e WCRO84_L005_map.err -o WCRO84_L005_map.out WCRO84_L005_map.sh | cut -d " " -f 4)
echo "WCRO84_L005_map.sh under the ID $WCRO84_L005 has been submitted"
WCRO84_L006=$(sbatch -A snic2017 -p core -n 20 -t 5-00:00:00 -J WCRO84_L006_map -e WCRO84_L006_map.err -o WCRO84_L006_map.out WCRO84_L006_map.sh | cut -d " " -f 4)
echo "WCRO84_L006_map.sh under the ID $WCRO84_L006 has been submitted"
WCRO84_mergeMarkDuplBQSR=$(sbatch --dependency=afterok:$WCRO84_L005:$WCRO84_L006 -A snic2017 -p core -n 1 -t 5-00:00:00 -J WCRO84_mergeMarkDuplBQSR -e WCRO84_mergeMarkDuplBQSR.err -o WCRO84_mergeMarkDuplBQSR.out WCRO84_mergeMarkDuplBQSR.sh | cut -d " " -f 4)
echo "WCRO84_mergeMarkDuplBQSR.sh under the ID $WCRO84_mergeMarkDuplBQSR has been submitted"
WCRO84_gVCF=$(sbatch --dependency=afterok:$WCRO84_mergeMarkDuplBQSR -A snic2017 -p core -n 1 -t 5-00:00:00 -J WCRO84_gVCF -e WCRO84_gVCF.err -o WCRO84_gVCF.out WCRO84_gVCF.sh | cut -d " " -f 4)
echo "WCRO84_gVCF.sh under the ID $WCRO84_gVCF has been submitted"
WCRO84_qualimap=$(sbatch --dependency=afterok:$WCRO84_mergeMarkDuplBQSR -A snic2017 -p core -n 1 -t 5-00:00:00 -J WCRO84_qualimap -e WCRO84_qualimap.err -o WCRO84_qualimap.out WCRO84_qualimap.sh | cut -d " " -f 4)
echo "WCRO84_qualimap.sh under the ID $WCRO84_qualimap has been submitted"
WCRO86_L005=$(sbatch -A snic2017 -p core -n 20 -t 5-00:00:00 -J WCRO86_L005_map -e WCRO86_L005_map.err -o WCRO86_L005_map.out WCRO86_L005_map.sh | cut -d " " -f 4)
echo "WCRO86_L005_map.sh under the ID $WCRO86_L005 has been submitted"
WCRO86_L007=$(sbatch -A snic2017 -p core -n 20 -t 5-00:00:00 -J WCRO86_L007_map -e WCRO86_L007_map.err -o WCRO86_L007_map.out WCRO86_L007_map.sh | cut -d " " -f 4)
echo "WCRO86_L007_map.sh under the ID $WCRO86_L007 has been submitted"
WCRO86_mergeMarkDuplBQSR=$(sbatch --dependency=afterok:$WCRO86_L005:$WCRO86_L007 -A snic2017 -p core -n 1 -t 5-00:00:00 -J WCRO86_mergeMarkDuplBQSR -e WCRO86_mergeMarkDuplBQSR.err -o WCRO86_mergeMarkDuplBQSR.out WCRO86_mergeMarkDuplBQSR.sh | cut -d " " -f 4)
echo "WCRO86_mergeMarkDuplBQSR.sh under the ID $WCRO86_mergeMarkDuplBQSR has been submitted"
WCRO86_gVCF=$(sbatch --dependency=afterok:$WCRO86_mergeMarkDuplBQSR -A snic2017 -p core -n 1 -t 5-00:00:00 -J WCRO86_gVCF -e WCRO86_gVCF.err -o WCRO86_gVCF.out WCRO86_gVCF.sh | cut -d " " -f 4)
echo "WCRO86_gVCF.sh under the ID $WCRO86_gVCF has been submitted"
WCRO86_qualimap=$(sbatch --dependency=afterok:$WCRO86_mergeMarkDuplBQSR -A snic2017 -p core -n 1 -t 5-00:00:00 -J WCRO86_qualimap -e WCRO86_qualimap.err -o WCRO86_qualimap.out WCRO86_qualimap.sh | cut -d " " -f 4)
echo "WCRO86_qualimap.sh under the ID $WCRO86_qualimap has been submitted"
Again, I assume and describe a cluster system with the Slurm Workload Manager, if your cluster is different, you may need to modify this script.
It works pretty simply. The script submits all mapping jobs (e.g. WCRO84_L00?_map.sh) as independent jobs, store their job ID in variables (e.g. $WCRO84_L005, $WCRO84_L006) and uses these IDs as dependencies for the subsequent WCRO84_mergeMarkDuplBQSR.sh job (--dependency=afterok:). In its turn, WCRO84_gVCF.sh uses the ID of WCRO84_mergeMarkDuplBQSR.sh as a dependency, etc.
Hardcoded parts
Most of the variables of this genomic variant calling pipeline can be changed with the command line options of reads-to-VCF.py. Let me remind you that you can see the available options with:
python reads-to-VCF.py -h
However, a few parts are hardcoded and you may need to modify them.
I also need to explain here that the script reads-to-VCF.py depends on sbatch.py module that is also located in my Github repository. I store all the functions in that module, so if you need to edit some hardcoded parts, you are most likely need to edit sbatch.py.
1. You may need to modify the functions writeMapBWAJob and writeMapStampyJob if your FASTQ file names don't follow this structure:
WCRO84_S1_L005_R1_001.fastq.gz
Alternatively, you can rename your files to fit this structure.
2. Uppmax Slurm outputs the job ID in a sentence: Submitted batch job 4724521 So, I had to use the command cut -d " " -f 4 to extract the ID. If your cluster simply outputs the ID, you can remove the cut command from the function writeSbatchScript.
3. Possibly, you also need to adjust MAX_FILE_HANDLES_FOR_READ_ENDS_MAP in the function writeMarkDuplJob according to your system. As a rule, it should be little smaller than the output of the ulimit -n command.
4. picard.jar is assumed to be located in the working directory. You can either edit the code (functions writeMapStampyJob , writeMarkDuplJob) to specify the full path to picard.jar, or copy picard.jar to your working directory before submitting the jobs.
Final thoughts
I hope you will use this genomic variant calling pipeline in your workflow. In my case of 82 dog samples with the mean coverage of x30, running the whole pipeline took little more than a week. Given that it is automatic and I was able to do some other work during this time, it is not a lot.
I know that such a pipeline can be made even more efficient and reliable with Nextflow and SnakeMake. But I found out about these workflow management systems after I wrote this pipeline. Moreover, I first thought to write this genomic variant calling pipeline in Apache Spark, but Spark is not mainstream yet and Spark version of the GATK is still in beta. So, I hope I will update this pipeline with a more advanced framework in the future.
If you have any questions or suggestions, feel free to email me.