
Heterozygotes as ambiguity characters. Mistakes you don’t want to make
Ambiguity characters are often used to code heterozygotes. However, using heterozygotes as ambiguity characters may bias many estimates.
The problem is that most software would use such heterozygous genotypes as uncertainty. This problem is very obvious but according to my experience, it frequently stays unnoticed. Let me elaborate little more below,
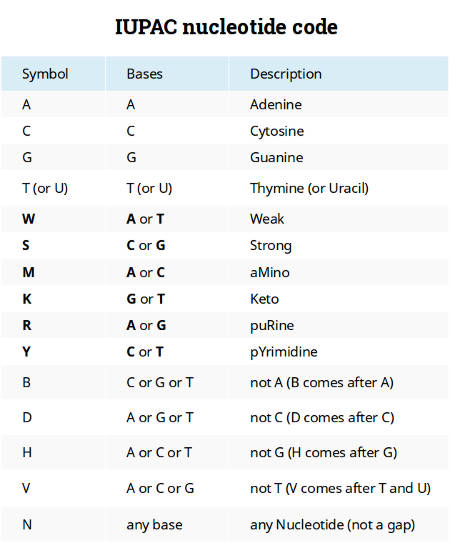
IUPAC nucleotide code
The current nucleic acid notation appeared a long time before the next-generation sequencing and whole genome data analyses. Characters A, C, G, and T were introduced to represent the four nucleotides of a DNA molecule. Ambiguity characters W, S, M, K, R, Y were proposed to code positions when there is some uncertainty between two nucleotides and B, D, H, V were used when there is only confidence that a position is not one of the four nucleotides. This coding system is known as the IUPAC nucleotide code.
It worked well in the early DNA sequencing era when scientist studied short haploid DNA sequences, and there is still no alternative today. All software uses this coding system.
Coding heterozygotes in the next-generation sequencing data
With the advent of next-generation sequencing when we started to work with genome-wide data of different ploidy, the same IUPAC coding system became being used in a little different way than it was originally designed. Many scientists, including myself, use ambiguity characters to code heterozygote sites in diploid organisms, e.g. the heterozygote A/T would be presented as W.
This is a convenient way to code because it allows keeping only one sequence per individual. This coding system also seems to be a reasonable way to make an input file for many software packages using formats like FASTA. Personally, I like to use the tab-delimited table format with the one-character code as shown below.
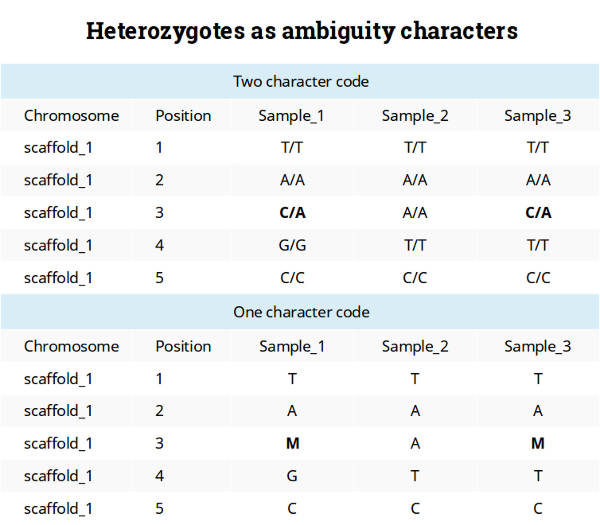
Heterozygotes as ambiguity characters may bias your analyses!
However, while it is known for sure that ambiguity characters in the data stand for two particular nucleotides and NOT for uncertainty, many software packages have been designed to analyze haploid sequences, and they would treat these heterozygote characters as ambiguity. Often such sites are just dropped out by a program.
This may introduce a substantial bias in such estimation as nucleotide diversity, absolute divergence and related population genetic statistics (e.g. Tajima's D). Heterozygotes as ambiguity characters may also affect branch lengths of a phylogenetic tree. The effect can be especially dramatic if there is a high level of heterozygosity in the data.
Nucleotide diversity (π) and divergence estimation (Dxy)
If heterozygous genotypes are coded as ambiguity characters and it is not properly accounted for, it mainly affects the nucleotide diversity estimation while all the other statistics just depend on it. For example, if nucleotide diversity is underestimated, the absolute divergence will also be underestimated.
By nucleotide diversity (know as π) I mean average pairwise differences per site between all samples representing one population. Absolute divergence (known as Dxy) is a similar estimate, but it is based on pairwise differences between representatives of different populations. Thus, they are similar estimates, but the former compares samples within groups, and the latter does it between groups.
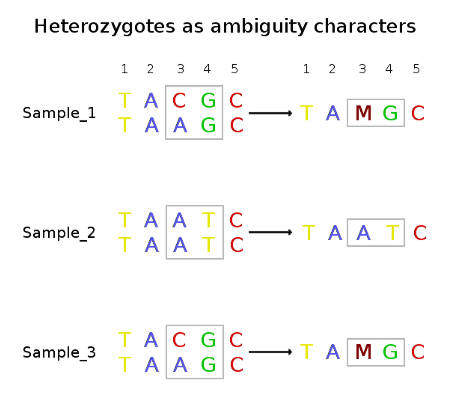
Let's have a look at the sequences of three diploid individuals above and estimate the divergence between them and nucleotide diversity of this whole sample. You can see that the sequences of Individual_A and Individual_C have heterozygous genotypes at position 3. Thus, each of these two individuals has only one sequence that differs from Individual_B in this position (C ≠ A and A = A ). On the other hand, at position 4 the difference between Individual_B and the other two individuals is in homozygous state and thus Individual_B has two different nucleotides at this position (G ≠ T and G ≠ T). Basically, when we estimate divergence between diploid sequences, we should compare each strand of the diploid DNA molecule.
If we calculate Dxy between these three individuals, we obtain the following values:
Individual_A vs. Individual_B: (2+2+1+1)/(5*4) = 0.3
Individual_A vs. Individual_C: (0+1+1+0)/(5*4) = 0.1
Individual_B vs. Individual_C: (2+2+1+1)/(5*4) = 0.3
The nucleotide diversity of this sample is just a mean of all pairwise differences between these six sequences, and it equals ~0.213.
Now let's have a look at the right part of the image, where heterozygous genotypes are coded as ambiguity characters. If a program discarded sites with ambiguity characters (position 3) and treated these data as haploid, it would analyze these data as there were only three sequences with four nucleotides and it would only find the difference at position 4. The divergence would be the following:
Individual_A vs. Individual_B: 1/4 = 0.25
Individual_A vs. Individual_C: 0/4 = 0.00
Individual_B vs. Individual_C: 1/4 = 0.25
The nucleotide diversity would be estimated as ~0.167, and it would be around 22% lower than the true value.
This bias can be very dramatic if the level of heterozygosity is very high and it probably can be negligible if the level of heterozygosity is very low. For example, I currently study Capsella bursa-pastrois, a species which has an elevated level of heterozygosity due to the hybrid origin (the alignment in the header of this post is from C. bursa-pastrois) and discarding the heterozygous sites results in the underestimation of both π and Dxy by more than ten times.
You could say, "But wait a minute, Individual_A and Individual_C are identical genetically, and it is correct that the difference between them is 0!". But you should not forget that in population genetics we are interested in alleles, not in individuals. And you would agree that the frequency of the allele A at position 2 and 3 in Individual_A and Individual_C are different (1 and 0.5, respectively).
Branch lengths of a phylogenetic tree
As far as I know, all phylogenetic programs usually do not use ambiguity characters as heterozygotes because there is yet no proper model to do so. So, depending on the distribution of heterozygosity in your data, this may have a substantial effect or may have no influence.
In my Master's thesis (see Publications), I did a phylogenetic analysis of Heliconius butterflies, and I observed that one of the species had a long branch length. However, when I pseudo-phased the data by random assignment of one of the alleles from heterozygous genotypes and thus allowed a phylogenetic program to use the information previously masked in the heterozygous sites, the branch length of this species was not that long anymore.
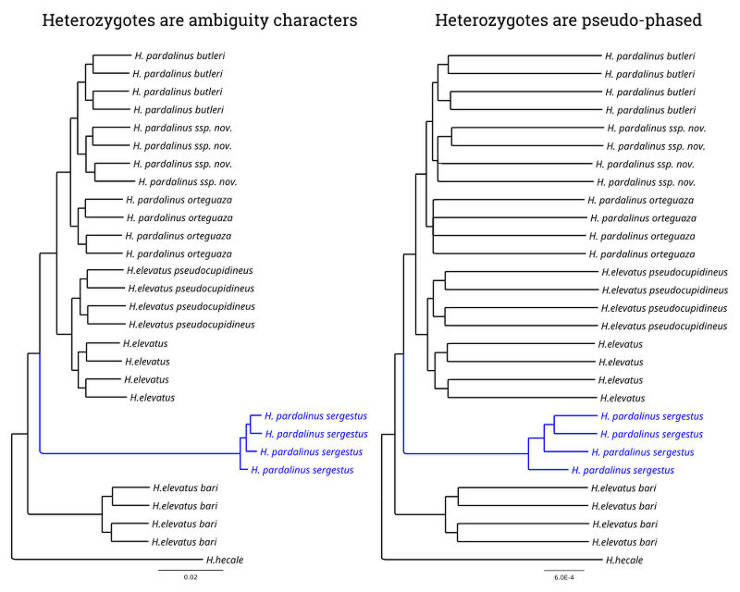
You can see that the branch length of H. pardalinus sergestus (blue) is substantially longer in non-phased data, where all heterozygotes are coded by ambiguity characters while in the pseudo-phased data, the length of H. pardalinus sergestus branch is even shorter than in the other taxa. Besides, you probably have also noticed that introducing heterozygous sites into the analysis substantially increased the relative length of the terminal branches. This is due to the elevation of genetic diversity unmasked by pseudo-phasing.
Software
Unfortunately, I cannot provide an extensive list of software which can or cannot use ambiguity characters as heterozygotes. Maybe I will make such a list later. For this post, I have only looked at the programs listed on the Wikipedia page about nucleotide diversity.
Among these programs, MEGA5 and VariScan discard ambiguity characters. Arlequin can handle heterozygous genotypes, but it requires that you provide actual haplotypes (real or pseudo-phased), and this is different from using ambiguity characters. DnaSP is the only program that correctly uses ambiguity characters as heterozygotes. You just need to use the option "Open Unphase/Genotype Data Files" during data loading.
I recommend to always check the performance of a program you are going to use. If a program does not recognize ambiguity characters as heterozygotes, you can pseudo-phase heterozygous genotypes and analyze sequences as haploids. I will explain how to do the pseudo-phasing in my next posts.
I would like to say that, of course, there is software designed specifically to analyze genomic data in the VCF format (e.g. VCFtools) where genotypes are presented by two or more alleles. But people often convert VCF files to some other formats and use ambiguity characters to code heterozygous genotypes and this may lead to the biases I describe.
Personally, I use custom python scripts to estimate π, Dxy, and other population genetic statistics, but this is a topic for another post.
If you have any questions or suggestions, feel free to email me.