Call germline Copy Number Variants with GATK in Snakemake
This pipeline calls germline copy number variants (CNV) with GATK 4 and Snakemake. It uses the cohort mode, so the CNV are inferred from all samples together.
I needed to call copy number variants (CNVs) in my dog dataset. I had different tools on my radar including Manata, LUMPY, CNVnator, and GenomeSTRiP. Among these tools, I liked Manata for its incredible speed. But it lacked the cohort mode calling which I thought was preferable for my population-level data. Only GenomeSTRiP had the cohort calling mode. I have not run GenomeSTRiP myself, but I talked to a person who tried it and he told me it was not the easiest tool to set up and run. I also recall GATK had a beta version that could call CNVs. Checking the GATK website revealed that this functionality has been released already. So, I decided to proceed with trusted GATK for calling germline copy number variants in my dataset.
The GATK documentation for this pipeline is in BETA for the moment of writing this post but it is enough to run the pipeline. I tested it and had no obvious problems. I am not going to describe each step of this pipeline in details as you can read about them. I will briefly list the steps and provide the Snakemake code to execute this pipeline.
Requirements
You will need GATK 4 in GATK Conda environment and Snakemake 4.
GATK Python environment
I run this pipeline with GATK 4.1.2.0. To call CNVs with GATK 4, you need to load a Python environment with gcnvkernel module. I use Conda installation for that:
conda env create -f /path/to/gatk/gatkcondaenv.yml
conda init bash # restart shell to take effect
conda activate gatk
Snakemake
I started writing this pipeline in Snakemake 5. I used recently introduced checkpoints to handle unknown output (see the scattering step below). But I encountered a problem which I was not able to fix. So, I downgraded to Snakemake 4.3.1 and used the older dynamic() function for scattering. Everything worked fine.
Steps to call copy number variants with GATK
These steps are described here only for a quick reference. For a detailed description of each step and options, read the GATK guide.
Bin intervals
PreprocessIntervals takes a reference fasta file as input and creates a binned interval lists. If you want to process only a subset of the genome, specify it with the option -L:
gatk --java-options "-Xmx8G" PreprocessIntervals \
-R canFam3.fa \
--padding 0 \
--bin-length 1000 \
-L chr35:100000-2000000 \
-imr OVERLAPPING_ONLY \
-O interval_chr35.interval_list
Bin size should negatively correlate with coverage, e.g. higher coverage data can have smaller bins. The default bin length of 1000 is recommended for 30x data.
Count reads per bin
This step counts reads overlapping each interval. It takes the interval list from the previous step and a BAM file as input and outputs a read counts table. The output can be in a human-readable TSV format (option --format TSV) or HDF5 (default) which is faster to process by GATK.
gatk --java-options "-Xmx8G" CollectReadCounts \
-R canFam3.fa \
-imr OVERLAPPING_ONLY \
-L interval_chr35.interval_list \
-I sample1.bam \
-O sample1_chr35.hdf5
OVERLAPPING_ONLY prevents the merging of abutting intervals as recommended by the GATK team.
Annotate and Filter intervals (Optional)
This step helps to remove problematic regions in the cohort calling mode. However, the pipeline should work fine without any interval filtering.
You can annotate intervals with GC content, mappability, and segmental duplication information:
gatk --java-options "-Xmx8G" AnnotateIntervals \
-R canFam3.fa \
-L interval_chr35.interval_list \
--mappability-track canFam3_mappability.bed.gz \
--segmental-duplication-track canFam3_segmental_duplication.bed.gz \
--interval-merging-rule OVERLAPPING_ONLY \
-O annotated_intervals_chr35.tsv
The information on mappability and segmental duplication need to be provided.
The GATK team recommends generating mappability with Umap and Bismap. I also used GEM to generate mappability.
To obtain segmental duplication information, I tried to run SEDEF and ASGART on the CamFam3 genome. Unfortunately, my attempts were unsuccessful: both programs crashed without a clear error message.
So, I annotated my data only with GC content and mappability.
Annotated intervals are then filtered based on tunable thresholds:
gatk --java-options "-Xmx8G" FilterIntervals \
-L interval_chr35.interval_list \
--annotated-intervals annotated_intervals_chr35.tsv \
-I sample1_chr35.hdf5 \
-I sample2_chr35.hdf5 \
--minimum-gc-content 0.1 \
--maximum-gc-content 0.9 \
--minimum-mappability 0.9 \
--maximum-mappability 1.0 \
--minimum-segmental-duplication-content 0.0 \
--maximum-segmental-duplication-content 0.5 \
--low-count-filter-count-threshold 5 \
--low-count-filter-percentage-of-samples 90.0 \
--extreme-count-filter-minimum-percentile 1.0 \
--extreme-count-filter-maximum-percentile 99.0 \
--extreme-count-filter-percentage-of-samples 90.0 \
--interval-merging-rule OVERLAPPING_ONLY \
-O gcfiltered_chr35.interval_list
Call contig ploidy
This step is needed to generate global baseline coverage and noise data for the subsequent steps:
gatk --java-options "-Xmx8G" DetermineGermlineContigPloidy \
-L interval_chr35.interval_list \
-I sample1_chr35.hdf5 \
-I sample2_chr35.hdf5 \
--contig-ploidy-priors ploidy_priors.tsv \
--output-prefix dog \
--interval-merging-rule OVERLAPPING_ONLY \
-O ploidy-calls_chr35
You need to provide ploidy prior probabilities. Here is an example of priors I used:
CONTIG_NAME PLOIDY_PRIOR_0 PLOIDY_PRIOR_1 PLOIDY_PRIOR_2 PLOIDY_PRIOR_3
chr35 0.01 0.01 0.97 0.01
chrX 0.01 0.49 0.49 0.01
If you have the information on the sex of your sample, it is advised to compare it with the ploidy call results.
Scatter intervals
GATK 4 utilizes a new approach for parallelization of processes that requires scattering your data. This step does exactly that. It splits the interval list into shards which can be processed in parallel. The results of these scattered processes are collected at the later step.
To scatter the intervals into ~5K intervals, run:
mkdir -p scatter_chr35
gatk --java-options "-Xmx8G" IntervalListTools \
--INPUT interval_chr35.interval_list \
--SUBDIVISION_MODE INTERVAL_COUNT \
--SCATTER_CONTENT 15000 \
--OUTPUT scatter_chr35
It is recommended to have at least ~10–50Mbp genomic coverage per scatter. So, scatters of ~15K with ~1K bins would have ~15Mb coverage.
Call copy number variants
This step detects both rare and common CNVs on a scattered shard:
gatk --java-options "-Xmx8G" GermlineCNVCaller \
--run-mode COHORT \
-L scatter_chr35/fragment/scattered.interval_list \
-I sample1_chr35.hdf5 \
-I sample2_chr35.hdf5 \
--contig-ploidy-calls ploidy-calls_chr35/dogs-calls \
--annotated-intervals annotated_intervals_chr35.tsv \
--output-prefix fragment \
--interval-merging-rule OVERLAPPING_ONLY \
-O cohort-calls_chr35
You need to run this command on each fragment produced by IntervalListTools from the Scattering step. This can be easely achived with Snakemake as you will see below.
To increase the sensitivity of calls, you need to fine-tune different parameters. For details visit this GATK page
Call copy number segments
This step collects the results from scattered shards and calls copy number state per sample for intervals and segments in the VCF format:
gatk --java-options "-Xmx8G" PostprocessGermlineCNVCalls \
--model-shard-path cohort-calls_chr35/frag_temp_0001_of_3-model \
--model-shard-path cohort-calls_chr35/frag_temp_0002_of_3-model \
--model-shard-path cohort-calls_chr35/frag_temp_0003_of_3-model \
--calls-shard-path cohort-calls_chr35/frag_temp_0001_of_3-calls \
--calls-shard-path cohort-calls_chr35/frag_temp_0002_of_3-calls \
--calls-shard-path cohort-calls_chr35/frag_temp_0003_of_3-calls \
--sequence-dictionary '/path/to/reference/canFam3.dict' \
--allosomal-contig chrX \
--contig-ploidy-calls ploidy-calls_chr35/dogs-calls \
--sample-index 0 \
--output-genotyped-intervals chr35_sample1_intervals_cohort.vcf.gz \
--output-genotyped-segments chr35_sample1_segments_cohort.vcf.gz
You need to provide a sample index with --sample-index. The first sample in your input list has index 0, the second one is 1, etc.
Here is an example of genotyped-segments in VCF:
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT sample1
13 chr35 100000 CNV_chr35_100000_309999 N <DEL>,<DUP> . . END=309999 GT:CN:NP:QA:QS:QSE:QSS 0:2:208:94:3077:98:136
14 chr35 310000 CNV_chr35_310000_311999 N <DEL>,<DUP> . . END=311999 GT:CN:NP:QA:QS:QSE:QSS 1:1:2:159:284:50:98
15 chr35 312000 CNV_chr35_312000_1999999 N <DEL>,<DUP> . . END=1999999 GT:CN:NP:QA:QS:QSE:QSS 0:2:1603:50:3077:131:50
GATK CNV pipeline in Snakemake
All the commands above can be executed as a distributed pipeline with Snakemake. For example, processing two chromosomes and two samples would look like this:
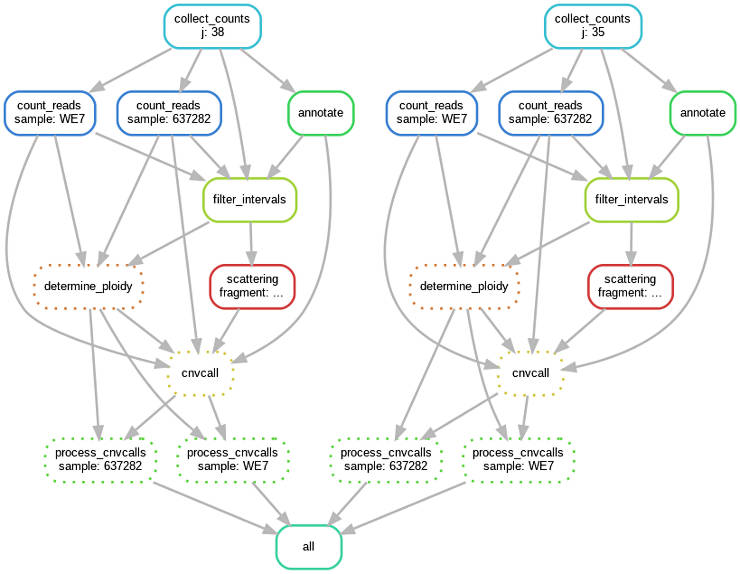
You can adapt the code below for your needs. Just change the list of input file names and chromosomes numbers.
SAMPLES, = glob_wildcards('/path/to/BAMs/{sample}_merged_markDupl_BQSR.bam')
CHRN = list(range(1, 39))
CHRN.append('X')
CHR = CHRN
REF = '/path/to/reference/canFam3.fa'
DICT = '/path/to/reference/canFam3.dict'
MAP = 'canFam3_mappability_150.merged.bed.gz'
SEGDUP = 'segmental_duplication.bed.gz'
rule all:
input:
expand('chr{j}_{sample}_intervals_cohort.vcf.gz', j=CHR, sample=SAMPLES),
expand('chr{j}_{sample}_segments_cohort.vcf.gz', j=CHR, sample=SAMPLES)
rule make_intervals:
input:
REF
params:
'chr{j}'
output:
'interval_chr{j}.interval_list'
shell:
'''
gatk --java-options "-Xmx8G" PreprocessIntervals \
-R {input} \
--padding 0 \
-L {params} \
-imr OVERLAPPING_ONLY \
-O {output}
'''
rule count_reads:
input:
ref = REF,
bam = '{sample}_merged_markDupl_BQSR.bam',
interval = 'interval_chr{j}.interval_list'
output:
'{sample}_chr{j}.hdf5'
shell:
'''
gatk --java-options "-Xmx8G" CollectReadCounts \
-R {input.ref} \
-imr OVERLAPPING_ONLY \
-L {input.interval} \
-I {input.bam} \
-O {output}
'''
rule annotate:
input:
ref = REF,
interval = 'interval_chr{j}.interval_list',
mappability = MAP,
segduplication = SEGDUP
output:
'annotated_intervals_chr{j}.tsv'
shell:
'''
gatk --java-options "-Xmx8G" AnnotateIntervals \
-R {input.ref} \
-L {input.interval} \
--mappability-track {input.mappability} \
--segmental-duplication-track {input.segduplication} \
--interval-merging-rule OVERLAPPING_ONLY \
-O {output}
'''
rule filter_intervals:
input:
interval = 'interval_chr{j}.interval_list',
annotated = 'annotated_intervals_chr{j}.tsv',
samples = expand('{sample}_{chromosome}.hdf5', sample=SAMPLES, chromosome='chr{j}'),
output:
'gcfiltered_chr{j}.interval_list'
params:
files = lambda wildcards, input: ' -I '.join(input.samples)
shell:
'''
gatk --java-options "-Xmx8G" FilterIntervals \
-L {input.interval} \
--annotated-intervals {input.annotated} \
-I {params.files} \
--interval-merging-rule OVERLAPPING_ONLY \
-O {output}
'''
rule determine_ploidy:
input:
interval = 'gcfiltered_chr{j}.interval_list',
samples = expand('{sample}_{chromosome}.hdf5', sample=SAMPLES, chromosome='chr{j}'),
prior = 'ploidy_priors.tsv',
params:
prefix = 'dogs',
files = lambda wildcards, input: ' -I '.join(input.samples)
output:
'ploidy-calls_chr{j}'
shell:
'''
gatk --java-options "-Xmx8G" DetermineGermlineContigPloidy \
-L {input.interval} \
-I {params.files} \
--contig-ploidy-priors {input.prior} \
--output-prefix {params.prefix} \
--interval-merging-rule OVERLAPPING_ONLY \
-O {output}
'''
rule scattering:
input:
interval = 'gcfiltered_chr{j}.interval_list'
output:
dynamic('scatter_chr{j}/{fragment}/scattered.interval_list')
params:
'scatter_chr{j}'
shell:
'''
mkdir -p {params} # needed because Snakemake fails creating this directory automatically
gatk --java-options "-Xmx8G" IntervalListTools \
--INPUT {input.interval} \
--SUBDIVISION_MODE INTERVAL_COUNT \
--SCATTER_CONTENT 15000 \
--OUTPUT {params}
'''
rule cnvcall:
input:
interval = 'scatter_chr{j}/{fragment}/scattered.interval_list',
sample = expand("{sample}_{chromosome}.hdf5", sample=SAMPLES, chromosome='chr{j}'),
annotated = 'annotated_intervals_chr{j}.tsv',
ploidy = 'ploidy-calls_chr{j}'
output:
modelf = "cohort-calls_chr{j}/frag_{fragment}-model",
callsf = "cohort-calls_chr{j}/frag_{fragment}-calls"
params:
outdir = 'cohort-calls_chr{j}',
outpref = 'frag_{fragment}',
files = lambda wildcards, input: " -I ".join(input.sample)
shell:
'''
gatk --java-options "-Xmx8G" GermlineCNVCaller \
--run-mode COHORT \
-L {input.interval} \
-I {params.files} \
--contig-ploidy-calls {input.ploidy}/dogs-calls \
--annotated-intervals {input.annotated} \
--output-prefix {params.outpref} \
--interval-merging-rule OVERLAPPING_ONLY \
-O {params.outdir}
'''
def sampleindex(sample):
index = SAMPLES.index(sample)
return index
rule process_cnvcalls:
input:
model = dynamic("cohort-calls_chr{j}/frag_{fragment}-model"),
calls = dynamic("cohort-calls_chr{j}/frag_{fragment}-calls"),
dict = DICT,
ploidy = 'ploidy-calls_chr{j}'
output:
intervals = 'chr{j}_{sample}_intervals_cohort.vcf.gz',
segments = 'chr{j}_{sample}_segments_cohort.vcf.gz'
params:
index = lambda wildcards: sampleindex(wildcards.sample),
modelfiles = lambda wildcards, input: " --model-shard-path ".join(input.model),
callsfiles = lambda wildcards, input: " --calls-shard-path ".join(input.calls)
shell:
'''
gatk --java-options "-Xmx8G" PostprocessGermlineCNVCalls \
--model-shard-path {params.modelfiles} \
--calls-shard-path {params.callsfiles} \
--sequence-dictionary {input.dict} \
--allosomal-contig chrX \
--contig-ploidy-calls {input.ploidy}/dogs-calls \
--sample-index {params.index} \
--output-genotyped-intervals {output.intervals} \
--output-genotyped-segments {output.segments}
'''
If you need to run Snakemake on a cluster, I explained how to do that previously.
Final thoughts
Although the documentation for copy number variants calling with GATK is in beta, it is sufficient to perform the CNV analysis. GATK is easy to install and it is reasonably fast. GATK now scatters the data during some steps to improve the efficacy. This approach is especially worthy if you run GATK on a Spark cluster. This is where large scale genomics is moving. However, if you do not have access to a full-scale Spark cluster, you can use GATK with this Snakemake pipeline on a cluster that has some job scheduler like SLURM, for example.
If you have any questions or suggestions, feel free to email me.