RNA-Seq STAR mapping with Snakemake
STAR mapping with Snakemake can save you a lot of time. STAR is a fast RNA-Seq aligner, whereas Snakemake provides automatic, reproducible, and scalable pipelining.
I have described my pipelines for genotype calling in both non-model and model organisms. I also showed how one can automate a genotype calling pipeline with automatically generated sbatch scripts that handle dependencies between jobs for the Slurm Workload Manager. I used a python script for that but I mentioned that probably it was not the most efficient way and using Nextflow or Snakemake would probably be a better option. I finally got my hands on Snakemake when I was working on my RNA-Seq mapping pipeline. You can read the description of this pipeline below and you can also get my Snakemake file at the end of this post to run this pipeline with your data.
RNA-Seq STAR mapping pipeline
There are many different mapping software for RNA-Seq data. The choice is always difficult. For example, I used stampy for RNA-seq mapping in my Capsella project. The reason behind this choice was that we performed an allele-specific expression analysis with the DNA count data as a null distribution. Therefore, to keep the consistency between the two datasets, I used the same aligner. In addition, stampy is not a bad aligner for RNA-Seq data and my favorite aligner for divergent reads in the genotyping pipeline.
However, for my current dog projects, I choose to use STAR aligner. It is a splicing aware aligner, and what is particularly important for large projects, it is one of the fastest aligners. I also use STAR in the multi-sample 2-pass mapping mode that better maps spliced reads (See STAR documentation).
The whole pipeline consists of STAR 2-pass alignment and reads counting with HTSeq:
-
Map reads to the reference genome (2-pass mode)
2.2. Collect the junctions information from all samples.
2.3. Use new junctions from all samples for the 2nd pass mapping.
All these STAR mapping steps can be automated with Snakemake as you will see below.
1. Index the reference genome
STAR needs to use its own index files during mapping. These index files are quite large. For example, for the dog reference genome, all STAR index files weight 23Gb, while the actual FASTA file is only 2.3Gb. But I believe that it is these large index files that allow STAR to perform alignment so fast.
So, to index the reference, you need to execute this code:
mkdir canFam3STAR
STAR --runThreadN 20 \
--runMode genomeGenerate \
--genomeDir canFam3STAR \
--genomeFastaFiles canFam3.fa \
--sjdbGTFfile canFam3.gtf \
--sjdbOverhang 100
I think these options are self-explanatory. --runThreadN indicates the number of cores to be used. --sjdbOverhang can be specified as ReadLength-1. You can also 100 which is recommended as a generally good value in the STAR documentation. canFam3 is the reference name for both FASTA and GTF file. You need to change this name for your reference in all commands below.
If you have only GFF annotation, you can convert GFF to GTF with Cufflinks:
gffread canFam3.1.92.gff3 -T -o canFam3.gtf
2. Run the mapping
You can run the standard 1-pass STAR mapping and the results should be good overall. However, given that STAR is very fast, running the 2-pass mode does not take too long and it can improve the mapping to novel junctions. Basically, you run the 1-pass STAR mapping to discover junctions information, then you collect and filter that information from all samples and run the 2-pass using that information.
2.1. Pass1 STAR mapping
The first pass of STAR mapping is a standard run that outputs an alignment and splice junction information.
mkdir Sample1_pass1
cd Sample1_pass1
STAR --runThreadN 20 \
--genomeDir /path/to/canFam3STAR \
--readFilesIn /path/to/Sample1_001_R1.fastq.gz,/path/to/Sample1_002_R1.fastq.gz /path/to/Sample1_001_R2.fastq.gz,/path/to/Sample1_002_R2.fastq.gz \
--readFilesCommand zcat \
--outSAMtype BAM Unsorted
Again, most of the options are self-explanatory. --readFilesCommand zcat is needed to extract gz compressed reads. --outSAMtype will output an unsorted BAM instead of a default SAM. This saves disk space. If you have your sample sequences in several lanes, you can list these files with comma separation in --readFilesIn as I did above.
This command will produce several output files, among which we are mostly interested in the splice junction information file SJ.out.tab that will be used in the next step. So. I discard the alignment BAM file because it takes too much disk space.
rm Sample1_pass1/Aligned.out.bam
2.2 Filter and collect the splicing information
To filter poorly supported junctions, I keep only the junctions that are supported by at least 3 uniquely mapped reads:
mkdir pass1SJ
for i in Sample*pass1/SJ.out.tab
do
awk '{ if ($7 >= 3) print $0}' $i > $i.filtered
mv $i.filtered pass1SJ/
done
rename SJ.out.tab.filtered SJ.filtered.tab pass1SJ/*.filtered
I think it is really difficult to verify splicing information. So, this filtering is rather subjective and can be skipped. I use it simply because of my gut feeling 🙂.
2.3 Pass2 STAR mapping
Now, we just execute almost the same mapping command as at step 2.1 but include add the information on the discovered splicing (--sjdbFileChrStartEnd). I also prefer to add read group information (--outSAMattrRGline) at this step. It is not necessary for reads counting but it may be useful in the future if I decide to use these STAR generated BAM files for other analyses.
mkdir Sample1pass2
cd Sample1pass2
STAR --runThreadN 20 \
--genomeDir /path/to/canFam3STAR \
--readFilesIn /path/to/Sample1_001_R1.fastq.gz,/path/to/Sample1_002_R1.fastq.gz /path/to/Sample1_001_R2.fastq.gz,/path/to/Sample1_002_R2.fastq.gz \
--readFilesCommand zcat \
--outSAMtype BAM SortedByCoordinate \
--outSAMattrRGline ID:Dog_MT2 \
--sjdbFileChrStartEnd Sample1_pass1.SJ.filtered.tab, ..., SampleN_pass1.SJ.filtered.tab \
3. Counting the number of reads per gene.
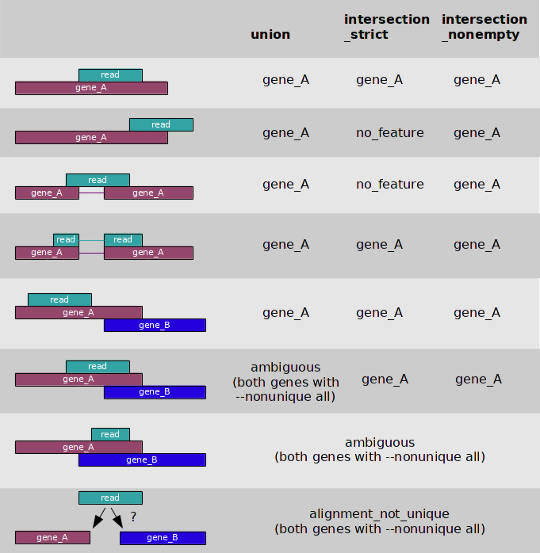
You can count the number of reads per gene on the fly during the STAR mapping if you provide it the option --quantMode GeneCounts. However, I prefer to count reads with htseq-count and use the option -m union to deal with overlapping features. You can see what the option -m union mean in the image below.
And here is the command I use:
htseq-count -m union -s no -t gene -i ID -f bam input.bam canFam3.gff &> output.log
grep gene output.log | sed 's/gene://g' > counts.csv
The second line extracts only the lines with counts per gene and cleans it by removing the string gene:.
The resulting file looks like this:
ENSCAFG00000000001 209
ENSCAFG00000000002 1
ENSCAFG00000000003 93
ENSCAFG00000000004 531
ENSCAFG00000000005 432
Finally, you can merge all files into one table:
for i in *csv; do sed -i "1igene\t$i" $i ; done # add column names
N=$(($(ls -l *.csv | wc -l)*2)) # count number of files
paste *csv | cut -f 1,$(seq -s, 2 2 $N) > all_HTSeq.csv # merge and keep only one column with gene names
This table will have the following format:
gene sample1 sample2 sample2
ENSCAFG00000000001 209 235 167
ENSCAFG00000000002 0 4 7
ENSCAFG00000000003 57 10 38
ENSCAFG00000000004 1243 1298 156
ENSCAFG00000000005 23 67 49
Snakemake STAR pipeline
All the commands above (except the last one that can be run locally) can be put together into a Snakemake file:
SAMPLES, = glob_wildcards('/path/to/fastq/{sample}_L001_R1.fastq.gz')
rule allout:
input:
directory('canFam3STAR'),
expand('{sample}_pass1/SJ.out.tab', sample=SAMPLES),
directory('SJ'),
expand('SJ/{sample}_pass1SJ.filtered.tab', sample=SAMPLES),
expand('{sample}_pass2/Aligned.sortedByCoord.out.bam', sample=SAMPLES),
expand('{sample}_HTSeq_union_gff3_no_gene_ID.log', sample=SAMPLES),
expand('{sample}_HTSeq.csv', sample=SAMPLES)
rule index:
input:
fa = 'canFam3.fa', # provide your reference FASTA file
gtf = 'canFam3.gtf' # provide your GTF file
output:
directory('canFam3STAR') # you can rename the index folder
threads: 20 # set the maximum number of available cores
shell:
'mkdir {output} && '
'STAR --runThreadN {threads} '
'--runMode genomeGenerate '
'--genomeDir {output} '
'--genomeFastaFiles {input.fa} '
'--sjdbGTFfile {input.gtf} '
'--sjdbOverhang 100'
rule pass1:
input:
R1L1 = 'fastq/{sample}/{sample}_L001_R1.fastq.gz', # may need adjustment if your fastq file name format is different
R1L2 = 'fastq/{sample}/{sample}_L002_R1.fastq.gz', # note each sample has 4 fastq files ~ 2 lanes per file
R2L1 = 'fastq/{sample}/{sample}_L001_R2.fastq.gz',
R2L2 = 'fastq/{sample}/{sample}_L002_R2.fastq.gz',
refdir = directory('canFam3STAR')
params:
outdir = '{sample}_pass1',
rmbam = '{sample}_pass1/Aligned.out.bam'
output:
'{sample}_pass1/SJ.out.tab'
threads: 20 # set the maximum number of available cores
shell:
'rm -rf {params.outdir} &&' # be careful with this. I don't know why, but Snakemake had problems without this cleaning.
'mkdir {params.outdir} && ' # snakemake had problems finding output files with --outFileNamePrefix, so I used this approach instead
'cd {params.outdir} && '
'STAR --runThreadN {threads} '
'--genomeDir {input.refdir} '
'--readFilesIn {input.R1L1},{input.R1L2} {input.R2L1},{input.R2L2} '
'--readFilesCommand zcat '
'--outSAMtype BAM Unsorted && rm {params.rmbam} && cd ..'
rule SJdir:
output:
directory('SJ')
threads: 1
shell:
'mkdir {output}'
rule filter:
input:
'{sample}_pass1/SJ.out.tab',
directory('SJ')
output:
'SJ/{sample}_pass1SJ.filtered.tab'
threads: 1
shell:
'''awk "{ { if (\$7 >= 3) print \$0 } }" {input[0]} > {input[0]}.filtered && '''
'mv {input[0]}.filtered {output}'
rule pass2:
input:
R1L1 = 'fastq/{sample}/{sample}_L001_R1.fastq.gz',
R1L2 = 'fastq/{sample}/{sample}_L002_R1.fastq.gz',
R2L1 = 'fastq/{sample}/{sample}_L001_R2.fastq.gz',
R2L2 = 'fastq/{sample}/{sample}_L002_R2.fastq.gz',
SJfiles = 'SJ/{sample}_pass1SJ.filtered.tab',
refdir = directory('canFam3STAR')
params:
outdir = '{sample}_pass2',
id = '{sample}'
output:
'{sample}_pass2/Aligned.sortedByCoord.out.bam'
threads: 20 # set the maximum number of available cores
shell:
'rm -rf {params.outdir} &&' # be careful with this. I don't know why, but Snakemake had problems without this cleaning.
'mkdir {params.outdir} && '
'cd {params.outdir} && '
'STAR --runThreadN {threads} '
'--genomeDir {input.refdir} '
'--readFilesIn {input.R1L1},{input.R1L2} {input.R2L1},{input.R2L2} '
'--readFilesCommand zcat '
'--outSAMtype BAM SortedByCoordinate '
'--sjdbFileChrStartEnd {input.SJfiles} '
'--outSAMattrRGline ID:{params.id} '
'--quantMode GeneCounts '
rule htseq:
input:
bam = '{sample}_pass2/Aligned.sortedByCoord.out.bam',
gff = 'canFam3.gff3'
output:
'{sample}_HTSeq_union_gff3_no_gene_ID.log',
'{sample}_HTSeq.csv'
threads: 1
shell:
'htseq-count -m union -s no -t gene -i ID -r pos -f bam {input.bam} {input.gff} &> {output[0]} && '
'grep ENS {output[0]} | sed "s/gene://g" > {output[1]}'
Read the comments within the code to find the line you need to change to adjust this Snakemake pipeline for your data.
Also, depending on your file location and Snakemake version, Snakemake may have problems finding files without the absolute path in file names. For example, instead of relative path fastq/{sample}_L001_R1.fastq.gz you may need to use the absolute path /home/username/RNA-Seq/fastq/{sample}_L001_R1.fastq.gz
Run Snakemake on a Slurm cluster (Uppmax)
I executed this Snakemake file on our Slurm cluster (Uppmax). To do that I created a Snakemake cluster config file cluster.yaml:
__default__:
account: snic2019-x-xxx
time: "00:01:00"
n: 1
partition: "core"
index:
time: "05:00:00"
n: 20
pass1:
time: "01:00:00"
n: 20
pass2:
time: "02:00:00"
n: 20
htseq:
time: "05:00:00"
This config file is used during Snakemake job submission with --cluster-config cluster.yaml.
I first run this pipeline in a dry mode with the --dryrun option:
snakemake -s Snakefile -j 100 --dryrun --cluster-config cluster.yaml --cluster "sbatch -A {cluster.account} -t {cluster.time} -p {cluster.partition} -n {cluster.n}"
If everything works fine in a dry mode, you can run this command in a regular mode from a login node of the server. However, I prefer to create a sbatch file (see below) and submit this command as a job which in turn will submit all other jobs as defined in the Snakemake file.
#!/bin/bash -l
#SBATCH -A snic2019-x-xxx
#SBATCH -p core
#SBATCH -n 1
#SBATCH -t 1-00:00:00
#SBATCH -J sbatchSnakefile
#SBATCH -e sbatchSnakefile.err
#SBATCH -o sbatchSnakefile.out
snakemake -s Snakefile -j 100 --cluster-config cluster.yaml --cluster "sbatch -A {cluster.account} -t {cluster.time} -p {cluster.partition} -n {cluster.n}"
Conclusion
Snakemake is a great tool and I am very happy that I have finally started using it. A combination of STAR speed and Snakemake workflow efficiency makes RNA-Seq mapping pipeline truly fast, robust, and error-safe. This pipeline has already saved me some time with my pilot RNA-Seq experiment and it will save even more time when my new RNA-Seq data will arrive.
I hope I will also update my genotype calling pipeline with Snakemake workflow soon.
If you have any questions or suggestions, feel free to email me.